
XTRAS
eXplainable TRanslational AI in Space
Principal Investigator: Prof. Dr. Stefan Simm, Universität Greifswald
Project Collaborators :
AI collaborators:
More About XTRAS
Milestones:
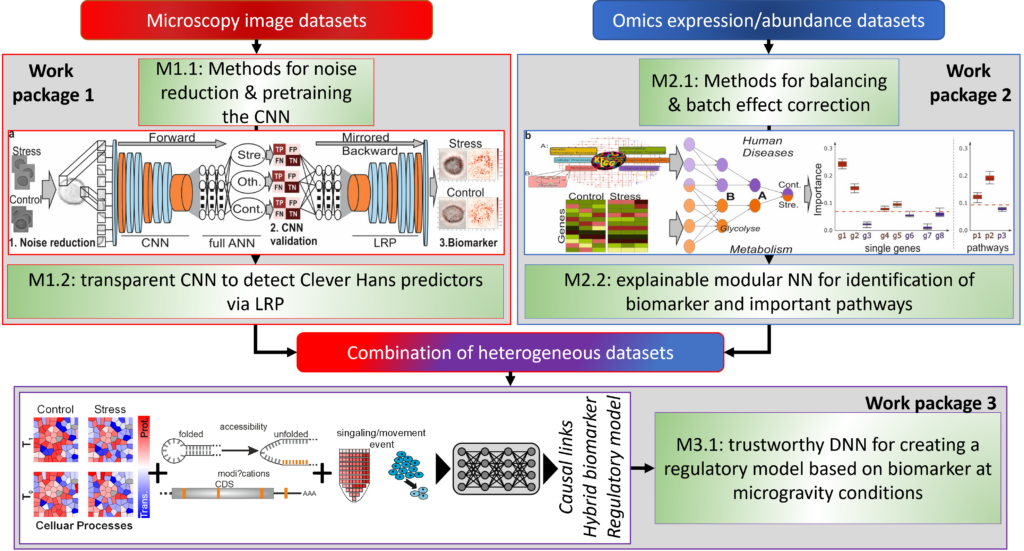
1) Development of a transparent CNN for Clever Hans predictors
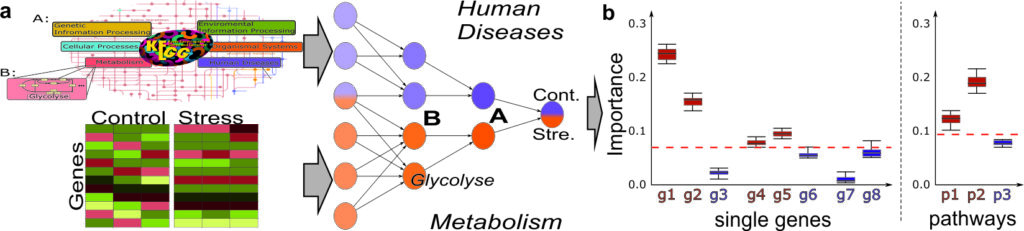
2) Modular NN for biomarkers and pathways
3) Development of a trustworthy DNN for the creation of a regulatory model
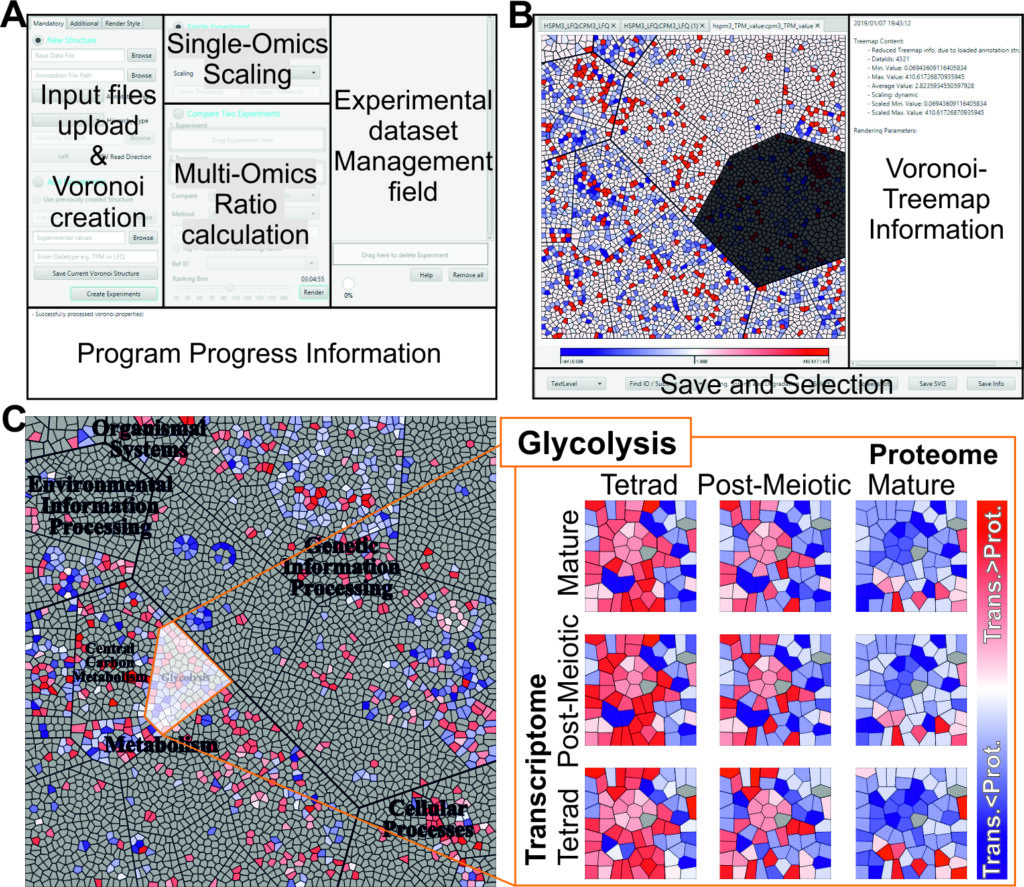
The central goal of the XTRAS single project is to implement explainable deep learning architectures with layerwise relevance propagation (LRP) to recognise biomarkers for microgravity influences in order to facilitate data analysis of high-throughput datasets in different organisms (plants and animals). Based on omics datasets, different levels of molecular interaction (transcriptome, proteome) and their interplay under microgravity in plants and animals will be used for classification. In addition, modular neural networks (NN) will be used to analyse the importance of individual genes and their interaction within important metabolic pathways, e.g. gravitropism in plants or metastasis in humans. In addition, images of roots from FLUMIAS microscope experiments will be automatically analyzed and preprocessed to identify different patterns for the differentiation of longitudinal microgravity experiments.
In both cases, methods for background noise reduction, confounding factor identification and processing and balancing steps will be developed that take into account the small number of replicates and their high variability in life science experiments. In parallel, an explainable AI method for linking heterogeneous data (microscope images, omics expression values, sequence/structure information) will be implemented to detect causal links for microgravity influences in human and plants.
Our Team

Prof. Dr. Stefan Simm

Jan Oldenburg
Publications
2024
Shanmugam, Thiruvenkadam; Chaturvedi, Palak; Streit, Deniz; Ghatak, Arindam; Bergelt, Thorsten; Simm, Stefan; Weckwerth, Wolfram; Schleiff, Enrico
Low dose ribosomal DNA P-loop mutation affects development and enforces autophagy in Arabidopsis. Journal Article
In: RNA biology, vol. 21, iss. 1, pp. 1-15, 2024, ISSN: 1555-8584.
@article{Shanmugam2024,
title = {Low dose ribosomal DNA P-loop mutation affects development and enforces autophagy in Arabidopsis.},
author = {Thiruvenkadam Shanmugam and Palak Chaturvedi and Deniz Streit and Arindam Ghatak and Thorsten Bergelt and Stefan Simm and Wolfram Weckwerth and Enrico Schleiff},
doi = {10.1080/15476286.2023.2298532},
issn = {1555-8584},
year = {2024},
date = {2024-01-01},
urldate = {2024-01-01},
journal = {RNA biology},
volume = {21},
issue = {1},
pages = {1-15},
abstract = {Arabidopsis contains hundreds of ribosomal DNA copies organized within the nucleolar organizing regions (NORs) in chromosomes 2 and 4. There are four major types of variants of rDNA, VAR1-4, based on the polymorphisms of 3' external transcribed sequences. The variants are known to be differentially expressed during plant development. We created a mutant by the CRISPR-Cas9-mediated excision of ~ 25 nt from predominantly NOR4 ribosomal DNA copies, obtaining mosaic mutational events on ~ 5% of all rDNA copies. The excised region consists of P-loop and Helix-82 segments of 25S rRNA. The mutation led to allelic, dosage-dependent defects marked by lateral root inhibition, reduced size, and pointy leaves, all previously observed for defective ribosomal function. The mutation in NOR4 led to dosage compensation from the NOR2 copies by elevated expression of VAR1 in mutants and further associated single-nucleotide variants, thus, resulting in altered rRNA sub-population. Furthermore, the mutants exhibited rRNA maturation defects specifically in the minor pathway typified by 32S pre-rRNA accumulation. Density-gradient fractionation and subsequent RT-PCR of rRNA analyses revealed that mutated copies were not incorporated into the translating ribosomes. The mutants in addition displayed an elevated autophagic flux as shown by the autophagic marker GFP-ATG8e, likely related to ribophagy.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2023
Bergquist, Timothy; Schaffter, Thomas; Yan, Yao; Yu, Thomas; Prosser, Justin; Gao, Jifan; Chen, Guanhua; Charzewski, Łukasz; Nawalany, Zofia; Brugere, Ivan; Retkute, Renata; Prusokas, Alidivinas; Prusokas, Augustinas; Choi, Yonghwa; Lee, Sanghoon; Choe, Junseok; Lee, Inggeol; Kim, Sunkyu; Kang, Jaewoo; Mooney, Sean D; Guinney, Justin; Consortium, Patient Mortality Prediction DREAM Challenge
Evaluation of crowdsourced mortality prediction models as a framework for assessing artificial intelligence in medicine. Journal Article
In: Journal of the American Medical Informatics Association : JAMIA, vol. 31, iss. 1, pp. 35-44, 2023, ISSN: 1527-974X.
@article{Bergquist2023,
title = {Evaluation of crowdsourced mortality prediction models as a framework for assessing artificial intelligence in medicine.},
author = {Timothy Bergquist and Thomas Schaffter and Yao Yan and Thomas Yu and Justin Prosser and Jifan Gao and Guanhua Chen and Łukasz Charzewski and Zofia Nawalany and Ivan Brugere and Renata Retkute and Alidivinas Prusokas and Augustinas Prusokas and Yonghwa Choi and Sanghoon Lee and Junseok Choe and Inggeol Lee and Sunkyu Kim and Jaewoo Kang and Sean D Mooney and Justin Guinney and Patient Mortality Prediction DREAM Challenge Consortium},
doi = {10.1093/jamia/ocad159},
issn = {1527-974X},
year = {2023},
date = {2023-01-01},
journal = {Journal of the American Medical Informatics Association : JAMIA},
volume = {31},
issue = {1},
pages = {35-44},
abstract = {OBJECTIVE Applications of machine learning in healthcare are of high interest and have the potential to improve patient care. Yet, the real-world accuracy of these models in clinical practice and on different patient subpopulations remains unclear. To address these important questions, we hosted a community challenge to evaluate methods that predict healthcare outcomes. We focused on the prediction of all-cause mortality as the community challenge question. MATERIALS AND METHODS Using a Model-to-Data framework, 345 registered participants, coalescing into 25 independent teams, spread over 3 continents and 10 countries, generated 25 accurate models all trained on a dataset of over 1.1 million patients and evaluated on patients prospectively collected over a 1-year observation of a large health system. RESULTS The top performing team achieved a final area under the receiver operator curve of 0.947 (95% CI, 0.942-0.951) and an area under the precision-recall curve of 0.487 (95% CI, 0.458-0.499) on a prospectively collected patient cohort. DISCUSSION Post hoc analysis after the challenge revealed that models differ in accuracy on subpopulations, delineated by race or gender, even when they are trained on the same data. CONCLUSION This is the largest community challenge focused on the evaluation of state-of-the-art machine learning methods in a healthcare system performed to date, revealing both opportunities and pitfalls of clinical AI.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Dunkel, Heiko; Wehrmann, Henning; Jensen, Lars R; Kuss, Andreas W; Simm, Stefan
MncR: Late Integration Machine Learning Model for Classification of ncRNA Classes Using Sequence and Structural Encoding. Journal Article
In: International journal of molecular sciences, vol. 24, iss. 10, 2023, ISSN: 1422-0067.
@article{Dunkel2023,
title = {MncR: Late Integration Machine Learning Model for Classification of ncRNA Classes Using Sequence and Structural Encoding.},
author = {Heiko Dunkel and Henning Wehrmann and Lars R Jensen and Andreas W Kuss and Stefan Simm},
doi = {10.3390/ijms24108884},
issn = {1422-0067},
year = {2023},
date = {2023-01-01},
journal = {International journal of molecular sciences},
volume = {24},
issue = {10},
abstract = {Non-coding RNA (ncRNA) classes take over important housekeeping and regulatory functions and are quite heterogeneous in terms of length, sequence conservation and secondary structure. High-throughput sequencing reveals that the expressed novel ncRNAs and their classification are important to understand cell regulation and identify potential diagnostic and therapeutic biomarkers. To improve the classification of ncRNAs, we investigated different approaches of utilizing primary sequences and secondary structures as well as the late integration of both using machine learning models, including different neural network architectures. As input, we used the newest version of RNAcentral, focusing on six ncRNA classes, including lncRNA, rRNA, tRNA, miRNA, snRNA and snoRNA. The late integration of graph-encoded structural features and primary sequences in our MncR classifier achieved an overall accuracy of >97%, which could not be increased by more fine-grained subclassification. In comparison to the actual best-performing tool ncRDense, we had a minimal increase of 0.5% in all four overlapping ncRNA classes on a similar test set of sequences. In summary, MncR is not only more accurate than current ncRNA prediction tools but also allows the prediction of long ncRNA classes (lncRNAs, certain rRNAs) up to 12.000 nts and is trained on a more diverse ncRNA dataset retrieved from RNAcentral.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2022
Gast, Martina; Nageswaran, Vanasa; Kuss, Andreas W; Tzvetkova, Ana; Wang, Xiaomin; Mochmann, Liliana H; Rad, Pegah Ramezani; Weiss, Stefan; Simm, Stefan; Zeller, Tanja; Voelzke, Henry; Hoffmann, Wolfgang; Völker, Uwe; Felix, Stefan B; Dörr, Marcus; Beling, Antje; Skurk, Carsten; Leistner, David-Manuel; Rauch, Bernhard H; Hirose, Tetsuro; Heidecker, Bettina; Klingel, Karin; Nakagawa, Shinichi; Poller, Wolfram C; Swirski, Filip K; Haghikia, Arash; Poller, Wolfgang
tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions. Journal Article
In: Cells, vol. 11, iss. 24, 2022, ISSN: 2073-4409.
@article{Gast2022,
title = {tRNA-like Transcripts from the NEAT1-MALAT1 Genomic Region Critically Influence Human Innate Immunity and Macrophage Functions.},
author = {Martina Gast and Vanasa Nageswaran and Andreas W Kuss and Ana Tzvetkova and Xiaomin Wang and Liliana H Mochmann and Pegah Ramezani Rad and Stefan Weiss and Stefan Simm and Tanja Zeller and Henry Voelzke and Wolfgang Hoffmann and Uwe Völker and Stefan B Felix and Marcus Dörr and Antje Beling and Carsten Skurk and David-Manuel Leistner and Bernhard H Rauch and Tetsuro Hirose and Bettina Heidecker and Karin Klingel and Shinichi Nakagawa and Wolfram C Poller and Filip K Swirski and Arash Haghikia and Wolfgang Poller},
doi = {10.3390/cells11243970},
issn = {2073-4409},
year = {2022},
date = {2022-01-01},
journal = {Cells},
volume = {11},
issue = {24},
abstract = {The evolutionary conserved NEAT1-MALAT1 gene cluster generates large noncoding transcripts remaining nuclear, while tRNA-like transcripts (mascRNA, menRNA) enzymatically generated from these precursors translocate to the cytosol. Whereas functions have been assigned to the nuclear transcripts, data on biological functions of the small cytosolic transcripts are sparse. We previously found NEAT1-/- and MALAT1-/- mice to display massive atherosclerosis and vascular inflammation. Here, employing selective targeted disruption of menRNA or mascRNA, we investigate the tRNA-like molecules as critical components of innate immunity. CRISPR-generated human ΔmascRNA and ΔmenRNA monocytes/macrophages display defective innate immune sensing, loss of cytokine control, imbalance of growth/angiogenic factor expression impacting upon angiogenesis, and altered cell-cell interaction systems. Antiviral response, foam cell formation/oxLDL uptake, and M1/M2 polarization are defective in ΔmascRNA/ΔmenRNA macrophages, defining first biological functions of menRNA and describing new functions of mascRNA. menRNA and mascRNA represent novel components of innate immunity arising from the noncoding genome. They appear as prototypes of a new class of noncoding RNAs distinct from others (miRNAs, siRNAs) by biosynthetic pathway and intracellular kinetics. Their NEAT1-MALAT1 region of origin appears as archetype of a functionally highly integrated RNA processing system.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Nath, Neetika; Simm, Stefan
Machine Learning Based Methods and Best Practices of microRNA-Target Prediction and Validation. Journal Article
In: Advances in experimental medicine and biology, vol. 1385, pp. 109-131, 2022, ISSN: 0065-2598.
@article{Nath2022,
title = {Machine Learning Based Methods and Best Practices of microRNA-Target Prediction and Validation.},
author = {Neetika Nath and Stefan Simm},
doi = {10.1007/978-3-031-08356-3_4},
issn = {0065-2598},
year = {2022},
date = {2022-01-01},
journal = {Advances in experimental medicine and biology},
volume = {1385},
pages = {109-131},
abstract = {Within the last years, more and more noncoding RNAs (ncRNAs) became the focal point to understand cell regulatory mechanisms because one class of ncRNAs, microRNAs (miRNAs), plays an essential role in translation repression or degradation of specific mRNAs and is implicated in disease etiology. miRNAs can serve as oncomiRs (oncogenic miRNAs) and tumor suppressor miRNAs, thus, miRNA therapeutics in clinical trials have become a vital component with respect to cancer treatment. To circumvent side-effects and allow an accurate effect it is crucial to accurately predict miRNAs and their mRNA targets. Over the last two decades, different approaches for miRNA prediction as well as miRNA target prediction have been developed and improved based on sequence and structure features. Nowadays, the abundance of high-throughput sequencing data and databases of miRNAs and miRNA targets from different species allow the training, testing, and validation of predicted miRNAs and miRNA targets with machine learning methods. This book chapter focuses on the important requirements for miRNA target prediction tools using ML like common features used for miRNA-binding site prediction. Furthermore, best practices for the prediction and validation of miRNA-mRNA targets are presented and set in the context of possible applications for cancer diagnosis and therapeutics.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Jagirdar, Gayatri; Elsner, Matthias; Scharf, Christian; Simm, Stefan; Borucki, Katrin; Peter, Daniela; Lalk, Michael; Methling, Karen; Linnebacher, Michael; Krohn, Mathias; Wolke, Carmen; Lendeckel, Uwe
In: Frontiers in genetics, vol. 13, pp. 931017, 2022, ISSN: 1664-8021.
@article{Jagirdar2022,
title = {Re-Expression of Tafazzin Isoforms in TAZ-Deficient C6 Glioma Cells Restores Cardiolipin Composition but Not Proliferation Rate and Alterations in Gene Expression.},
author = {Gayatri Jagirdar and Matthias Elsner and Christian Scharf and Stefan Simm and Katrin Borucki and Daniela Peter and Michael Lalk and Karen Methling and Michael Linnebacher and Mathias Krohn and Carmen Wolke and Uwe Lendeckel},
doi = {10.3389/fgene.2022.931017},
issn = {1664-8021},
year = {2022},
date = {2022-01-01},
journal = {Frontiers in genetics},
volume = {13},
pages = {931017},
abstract = {Tafazzin-an acyltransferase-is involved in cardiolipin (CL) remodeling. CL is associated with mitochondrial function, structure and more recently with cell proliferation. Various tafazzin isoforms exist in humans. The role of these isoforms in cardiolipin remodeling is unknown. Aim of this study was to investigate if specific isoforms like Δ5 can restore the wild type phenotype with respect to CL composition, cellular proliferation and gene expression profile. In addition, we aimed to determine the molecular mechanism by which tafazzin can modulate gene expression by applying promoter analysis and (Ingenuity Pathway Analyis) IPA to genes regulated by TAZ-deficiency. Expression of Δ5 and rat full length TAZ in C6-TAZ- cells could fully restore CL composition and-as proven for Δ5-this is naturally associated with restoration of mitochondrial respiration. A similar restoration of CL-composition could not be observed after re-expression of an enzymatically dead full-length rat TAZ (H69L; TAZMut). Re-expression of only rat full length TAZ could restore proliferation rate. Surprisingly, the Δ5 variant failed to restore wild-type proliferation. Further, as expected, re-expression of the TAZMut variant completely failed to reverse the gene expression changes, whereas re-expression of the TAZ-FL variant largely did so and the Δ5 variant to somewhat less extent. Very likely TAZ-deficiency provokes substantial long-lasting changes in cellular lipid metabolism which contribute to changes in proliferation and gene expression, and are not or only very slowly reversible.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Gather, Leonie; Nath, Neetika; Falckenhayn, Cassandra; Oterino-Sogo, Sergio; Bosch, Thomas; Wenck, Horst; Winnefeld, Marc; Grönniger, Elke; Simm, Stefan; Siracusa, Annette
Macrophages Are Polarized toward an Inflammatory Phenotype by their Aged Microenvironment in the Human Skin. Journal Article
In: The Journal of investigative dermatology, vol. 142, iss. 12, pp. 3136-3145.e11, 2022, ISSN: 1523-1747.
@article{Gather2022,
title = {Macrophages Are Polarized toward an Inflammatory Phenotype by their Aged Microenvironment in the Human Skin.},
author = {Leonie Gather and Neetika Nath and Cassandra Falckenhayn and Sergio Oterino-Sogo and Thomas Bosch and Horst Wenck and Marc Winnefeld and Elke Grönniger and Stefan Simm and Annette Siracusa},
doi = {10.1016/j.jid.2022.06.023},
issn = {1523-1747},
year = {2022},
date = {2022-01-01},
journal = {The Journal of investigative dermatology},
volume = {142},
issue = {12},
pages = {3136-3145.e11},
abstract = {Aging of the skin is accompanied by cellular as well as tissue environmental changes, ultimately reducing the ability of the tissue to regenerate and adequately respond to external stressors. Macrophages are important gatekeepers of tissue homeostasis, and it has been reported that their number and phenotype change during aging in a site-specific manner. How aging affects human skin macrophages and what implications this has for the aging process in the tissue are still not fully understood. Using single-cell RNA-sequencing analysis, we show that there is at least a 50% increase of macrophages in human aged skin, which appear to have developed from monocytes and exhibit more proinflammatory M1-like characteristics. In contrast, the cell-intrinsic ability of aged monocytes to differentiate into M1 macrophages was reduced. Using coculture experiments with aged dermal fibroblasts, we show that it is the aged microenvironment that drives a more proinflammatory phenotype of macrophages in the skin. This proinflammatory M1-like phenotype in turn negatively influenced the expression of extracellular matrix proteins by fibroblasts, emphasizing the impact of the aged macrophages on the skin phenotype.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Fester, Niclas; Zielonka, Elisabeth; Goldmann, Jakob; Frombach, Ann-Sophie; Müller-Kuller, Uta; Gutfreund, Niklas; Riegel, Kristina; Smits, Jos G A; Schleiff, Enrico; Rajalingam, Krishnaraj; Zhou, Huiqing; Simm, Stefan; Dötsch, Volker
Enhanced pro-apoptosis gene signature following the activation of TAp63α in oocytes upon γ irradiation. Journal Article
In: Cell death & disease, vol. 13, iss. 3, pp. 204, 2022, ISSN: 2041-4889.
@article{Fester2022,
title = {Enhanced pro-apoptosis gene signature following the activation of TAp63α in oocytes upon γ irradiation.},
author = {Niclas Fester and Elisabeth Zielonka and Jakob Goldmann and Ann-Sophie Frombach and Uta Müller-Kuller and Niklas Gutfreund and Kristina Riegel and Jos G A Smits and Enrico Schleiff and Krishnaraj Rajalingam and Huiqing Zhou and Stefan Simm and Volker Dötsch},
doi = {10.1038/s41419-022-04659-2},
issn = {2041-4889},
year = {2022},
date = {2022-01-01},
journal = {Cell death & disease},
volume = {13},
issue = {3},
pages = {204},
abstract = {Specialized surveillance mechanisms are essential to maintain the genetic integrity of germ cells, which are not only the source of all somatic cells but also of the germ cells of the next generation. DNA damage and chromosomal aberrations are, therefore, not only detrimental for the individual but affect the entire species. In oocytes, the surveillance of the structural integrity of the DNA is maintained by the p53 family member TAp63α. The TAp63α protein is highly expressed in a closed and inactive state and gets activated to the open conformation upon the detection of DNA damage, in particular DNA double-strand breaks. To understand the cellular response to DNA damage that leads to the TAp63α triggered oocyte death we have investigated the RNA transcriptome of oocytes following irradiation at different time points. The analysis shows enhanced expression of pro-apoptotic and typical p53 target genes such as CDKn1a or Mdm2, concomitant with the activation of TAp63α. While DNA repair genes are not upregulated, inflammation-related genes become transcribed when apoptosis is initiated by activation of STAT transcription factors. Furthermore, comparison with the transcriptional profile of the ΔNp63α isoform from other studies shows only a minimal overlap, suggesting distinct regulatory programs of different p63 isoforms.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2020
Keller, Mario; Schleiff, Enrico; Simm, Stefan
miRNAs involved in transcriptome remodeling during pollen development and heat stress response in Solanum lycopersicum. Journal Article
In: Scientific reports, vol. 10, iss. 1, pp. 10694, 2020, ISSN: 2045-2322.
@article{Keller2020,
title = {miRNAs involved in transcriptome remodeling during pollen development and heat stress response in Solanum lycopersicum.},
author = {Mario Keller and Enrico Schleiff and Stefan Simm},
doi = {10.1038/s41598-020-67833-6},
issn = {2045-2322},
year = {2020},
date = {2020-01-01},
journal = {Scientific reports},
volume = {10},
issue = {1},
pages = {10694},
abstract = {Cellular transitions during development and stress response depend on coordinated transcriptomic and proteomic alterations. Pollen is particular because its development is a complex process that includes meiotic and mitotic divisions which causes a high heat sensitivity of these cells. Development and stress response are accompanied by a reprogramming of the transcriptome, e.g. by post-transcriptional regulation via miRNAs. We identified known and potentially novel miRNAs in the transcriptome of developing and heat-stressed pollen of Solanum lycopersicum (tomato). The prediction of target mRNAs yielded an equal number of predicted target-sites in CDS and 3'UTR regions of target mRNAs. The result enabled the postulation of a possible link between miRNAs and a fine-tuning of transcription factor abundance during pollen development. miRNAs seem to play a role in the pollen heat stress response as well. We identified several heat stress transcription factors and heat shock proteins as putative targets of miRNAs in response to heat stress, thereby placing these miRNAs as important elements of thermotolerance. Moreover, for members of the AP2, SBP and ARF family members we could predict a miRNA-mediated regulation during development via the miR172, mir156 and mir160-family strengthening the current concept of a cross-connection between development and stress response in plants.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
Hu, Yangjie; Fragkostefanakis, Sotirios; Schleiff, Enrico; Simm, Stefan
Transcriptional Basis for Differential Thermosensitivity of Seedlings of Various Tomato Genotypes. Journal Article
In: Genes, vol. 11, iss. 6, 2020, ISSN: 2073-4425.
@article{Hu2020,
title = {Transcriptional Basis for Differential Thermosensitivity of Seedlings of Various Tomato Genotypes.},
author = {Yangjie Hu and Sotirios Fragkostefanakis and Enrico Schleiff and Stefan Simm},
doi = {10.3390/genes11060655},
issn = {2073-4425},
year = {2020},
date = {2020-01-01},
journal = {Genes},
volume = {11},
issue = {6},
abstract = {Transcriptional reprograming after the exposure of plants to elevated temperatures is a hallmark of stress response which is required for the manifestation of thermotolerance. Central transcription factors regulate the stress survival and recovery mechanisms and many of the core responses controlled by these factors are well described. In turn, pathways and specific genes contributing to variations in the thermotolerance capacity even among closely related plant genotypes are not well defined. A seedling-based assay was developed to directly compare the growth and transcriptome response to heat stress in four tomato genotypes with contrasting thermotolerance. The conserved and the genotype-specific alterations of mRNA abundance in response to heat stress were monitored after exposure to three different temperatures. The transcripts of the majority of genes behave similarly in all genotypes, including the majority of heat stress transcription factors and heat shock proteins, but also genes involved in photosynthesis and mitochondrial ATP production. In turn, genes involved in hormone and RNA-based regulation, such as auxin- and ethylene-related genes, or transcription factors like HsfA6b, show a differential regulation that associates with the thermotolerance pattern. Our results provide an inventory of genes likely involved in core and genotype-dependent heat stress response mechanisms with putative role in thermotolerance in tomato seedlings.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2019
Berz, Jannik; Simm, Stefan; Schuster, Sebastian; Scharf, Klaus-Dieter; Schleiff, Enrico; Ebersberger, Ingo
HEATSTER: A Database and Web Server for Identification and Classification of Heat Stress Transcription Factors in Plants. Journal Article
In: Bioinformatics and biology insights, vol. 13, pp. 1177932218821365, 2019, ISSN: 1177-9322.
@article{Berz2019,
title = {HEATSTER: A Database and Web Server for Identification and Classification of Heat Stress Transcription Factors in Plants.},
author = {Jannik Berz and Stefan Simm and Sebastian Schuster and Klaus-Dieter Scharf and Enrico Schleiff and Ingo Ebersberger},
url = {http://journals.sagepub.com/doi/10.1177/1177932218821365 http://www.ncbi.nlm.nih.gov/pubmed/30670918 http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC6327235},
doi = {10.1177/1177932218821365},
issn = {1177-9322},
year = {2019},
date = {2019-01-01},
journal = {Bioinformatics and biology insights},
volume = {13},
pages = {1177932218821365},
abstract = {Heat stress transcription factors (HSFs) regulate transcriptional response to a large number of environmental influences, such as temperature fluctuations and chemical compound applications. Plant HSFs represent a large and diverse gene family. The HSF members vary substantially both in gene expression patterns and molecular functions. HEATSTER is a web resource for mining, annotating, and analyzing members of the different classes of HSFs in plants. A web-interface allows the identification and class assignment of HSFs, intuitive searches in the database and visualization of conserved motifs, and domains to classify novel HSFs.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2018
Keller, Mario; Consortium, SPOT-ITN; Simm, Stefan
The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen. Journal Article
In: BMC genomics, vol. 19, iss. 1, pp. 447, 2018, ISSN: 1471-2164.
@article{Keller2018,
title = {The coupling of transcriptome and proteome adaptation during development and heat stress response of tomato pollen.},
author = {Mario Keller and SPOT-ITN Consortium and Stefan Simm},
doi = {10.1186/s12864-018-4824-5},
issn = {1471-2164},
year = {2018},
date = {2018-01-01},
journal = {BMC genomics},
volume = {19},
issue = {1},
pages = {447},
abstract = {BACKGROUND Pollen development is central for plant reproduction and is assisted by changes of the transcriptome and proteome. At the same time, pollen development and viability is largely sensitive to stress, particularly to elevated temperatures. The transcriptomic and proteomic changes during pollen development and of different stages in response to elevated temperature was targeted to define the underlying molecular principles. RESULTS The analysis of the transcriptome and proteome of Solanum lycopersicum pollen at tetrad, post-meiotic and mature stage before and after heat stress yielded a decline of the transcriptome but an increase of the proteome size throughout pollen development. Comparison of the transcriptome and proteome led to the discovery of two modes defined as direct and delayed translation. Here, genes of distinct functional processes are under the control of direct and delayed translation. The response of pollen to elevated temperature occurs rather at proteome, but not as drastic at the transcriptome level. Heat shock proteins, proteasome subunits, ribosomal proteins and eukaryotic initiation factors are most affected. On the example of heat shock proteins we demonstrate a decoupling of transcript and protein levels as well as a distinct regulation between the developmental stages. CONCLUSIONS The transcriptome and proteome of developing pollen undergo drastic changes in composition and quantity. Changes at the proteome level are a result of two modes assigned as direct and delayed translation. The response of pollen to elevated temperature is mainly regulated at the proteome level, whereby proteins related to synthesis and degradation of proteins are most responsive and might play a central role in the heat stress response of pollen.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}
2017
Keller, M.; Hu, Y.; Mesihovic, A.; Fragkostefanakis, S.; Schleiff, E.; Simm, S.
Alternative splicing in tomato pollen in response to heat stress Journal Article
In: DNA Research, vol. 24, iss. 2, 2017, ISSN: 17561663.
@article{Keller2017,
title = {Alternative splicing in tomato pollen in response to heat stress},
author = {M. Keller and Y. Hu and A. Mesihovic and S. Fragkostefanakis and E. Schleiff and S. Simm},
doi = {10.1093/dnares/dsw051},
issn = {17561663},
year = {2017},
date = {2017-01-01},
journal = {DNA Research},
volume = {24},
issue = {2},
abstract = {© The Author 2017. Alternative splicing (AS) is a key control mechanism influencing signal response cascades in different developmental stages and under stress conditions. In this study, we examined heat stress (HS)-induced AS in the heat sensitive pollen tissue of two tomato cultivars. To obtain the entire spectrum of HS-related AS, samples taken directly after HS and after recovery were combined and analysed by RNA-seq. For nearly 9,200 genes per cultivar, we observed at least one AS event under HS. In comparison to control, for one cultivar we observed 76% more genes with intron retention (IR) or exon skipping (ES) under HS. Furthermore, 2,343 genes had at least one transcript with IR or ES accumulated under HS in both cultivars. These genes are involved in biological processes like protein folding, gene expression and heat response. Transcriptome assembly of these genes revealed that most of the alternative spliced transcripts possess truncated coding sequences resulting in partial or total loss of functional domains. Moreover, 141 HS specific and 22 HS repressed transcripts were identified. Further on, we propose AS as layer of stress response regulating constitutively expressed genes under HS by isoform abundance.},
keywords = {},
pubstate = {published},
tppubtype = {article}
}

Send Us A Message
Whether you are curious about the our work or want to collaborate with us, reach out to us. We are listening.